Болезнь Альцгеймера
Это весьма широко распространенное нейродегенеративное заболевание (по данным американских патологов на долю болезни Альцгеймера приходится до 60% всех случаев деменции в странах Европы и Северной Америки) получило свое название благодаря трудам известного немецкого патолога и патоморфолога Алоиза Альцгеймера (Альцхаймера).
А. Альцгеймер получил образование в Вюрцбурге и в Берлине. После защиты докторской диссертации в Вюрцбурге работал во Франкфурте-на-Майне. Затем по приглашению известного немецкого психиатра Э. Крепелина - в его клинике, сначала в Гейдельберге, а затем в Мюнхене. Его коллегами по работе были выдающиеся патологи и гистологи Ф. Ниссль и К. Бродман.В 1906 году на съезде психиатров в Тюбингеме А. Альцгеймер сообщил об открытой им новой болезни - особом виде старческого слабоумия, начинавшегося значительно раньше, чем обычное сенильное слабоумие.
По предложению Э. Крепелина (в 1912 году) болезнь получила название по имени автора (современное название -деменция Альцгеймеровского типа)
Болезнь Альцгеймера отличается медленным, но неотвратимым прогрессированием. От момента заболевания до смерти пациента проходит от 5 до 15 лет. В частности, по данным американских клиницистов у сорокового президента США Рональда Рейгана первые проявления болезни Альцгеймера были зарегистрированы еще в 1988 году, в конце его второго президентского срока. Скончался же Р.Рейган в 2004 году в возрасте 93 лет.
Клинические проявления болезни Альцгеймера связаны, прежде всего, с постепенно прогрессирующими нарушениями памяти, по принципу «от настоящего к прошлому». У больных нарушается внимание, они становятся неспособными сосредоточится на какой либо проблеме, неуклонное нарастание симптомов прогрессирующей деменции постепенно приводит к нарушению процессов мышления и способности к обучению, дезориентация во времени и пространстве, затруднения при подборе слов, трудности в общении, изменения личности.
Со временем больные полностью утрачивает способность к самообслуживанию, становятся беспомощными, требуют постоянного наблюдения со стороны родственников или медицинских работников. Смерть наступает при явлениях острой сердечной недостаточности или нарушений дыхания, вызванных параличом дыхательногоцентра.
Не существует каких либо специфических тестов для установления диагноза болезни Альцгеймера. Окончательно и достоверно диагноз болезни Альцгеймера может быть поставлен только по результатам патологоанатомического исследования мозга погибшего пациента. При предварительной постановке диагноза этого заболевания рекомендуется учитывать следующие облигатные признаки:
- наличие синдрома деменции;
- малозаметное начало и неуклонное прогрессирование синдрома деменции, наличие, по крайней мере, одного или нескольких когнитивных нарушений: афазии, апраксии, агнозии;
- заметное снижение социальной и профессиональной адаптации в результате прогрессирования деменции и нарушения когнитивных функций;
- анамнестические сведения и данные клинических исследований, исключающие наличие какого либо определенного психического заболевания или повреждения ЦНС.
Клинически определяется четыре стадии в развитии болезни Альцгеймера: стадия сомнительной деменции, стадия мягкой деменции, стадия умеренной деменции, стадия тяжелой деменции. Таким образом, в основу определения стадийности развития заболевания положены данные о постепенно нарастающих нарушениях памяти, когнитивных нарушениях и изменениях в уровне профессиональной и социальной адаптации.
Мы уже указывали, что, по мнению А.Альцгеймера, описанное им заболевание наступает в более ранние возрастные сроки, чем известное старческое слабоумие, связанное, как правило, с атеросклеротическими изменениями сосудов головного мозга и следующими за ними метаболическими нарушениями. Это обстоятельство лежит в основе современной классификации болезни Альцгеймера:
- пресенильная деменция альцгеймеровского типа (заболевание в возрасте до 65 лет; возможны случаи заболевания в 30 - 35 лет);
- сенильная деменция альцгеймеровского типа (заболевание в возрасте после 65 лет);
- деменция смешанного типа (атипичная болезнь Альцгеймера; сочетание признаков классической болезни Альцгеймера и сосудистой деменции).
Патоморфологическая картина болезни Альцгеймера характеризуется атрофией коры головного мозга, сужением извилин, углублением борозд, а также уменьшением объема и массы головного мозга. Желудочки мозга расширены. Гистологические исследования показывают значительное уменьшение числа нейронов коры мозга, гиппокампа и многих подкорковых образований. Отмечается дегенерация синапсов и дендритов, а также белого вещества головного мозга. Наиболее ярко эти изменения проявляются в медиобазальных отделах коры больших полушарий, в гиппокампе, височной коре и базальных ганглиях. Характерными патоморфологическими изменениями головного мозга при болезни Альцгеймера являются диффузно распространенные так называемые «сенильные амилоидные бляшки» и «нейрофибриллярные клубки». При этом амилоидные бляшки располагаются экстрацеллюляроно, а нейрофибриллярные клубки образуются в цитоплазме нейронов. С момента описания сенильных бляшек в головном мозгу людей, погибших от болезни Альцгеймера, и вплоть до сегодняшнего времени большинство исследователей считает их основной причиной возникновения этой патологии. По мнению патоморфологов амилоидные сенильные бляшки способствуют дегенерации синаптических образований, нарушают метаболизм нейронов, стимулируют развитие воспалительного процесса, а также увеличивают концентрацию ионов Са++ в нейроплазме. В результате пораженные нейроны погибают как путем некроза, так и апоптоза. Внутриклеточные интранейронные сплетения состоят из агрегатов гиперфосфорилированного белка - тау-протеина, который в норме стабилизирует входящие в состав цитоскелета микротрубочки («ми- кротуболи»), основная функция которых заключается в транспорте питательных и регуляционных веществ (так называемых «трофогенов») из нейроплазмы по микро- туболям аксонов к иннервируемым клеточным структурам и обратно, в цитоплазму нейронов. Следует иметь в виду, что нейрофибриллярные включения отмечаются и при ряде других нейродегенеративных заболеваний и, таким образом, не выступают как определяющий морфологический критерий болезни Альцгеймера.
Более того, некоторые патоморфологи считают, что появление этих включений являются следствием, а не причиной гибели нейронов.Каковы же конкретные патогенетические механизмы, лежащие в основе болезни Альцгеймера?
Для того чтобы разобраться в этой проблеме мы должны, прежде всего, разобраться в патогенезе ранних («семейных») форм этого заболевания, безусловно имеющих в своей основе вполне определенные генетические причины.
В настоящее время идентифицированы три гена, мутации которых приводят к возникновению наследственных (аутосомно-доминантных) форм болезни Альцгеймера, характеризующихся ранним развитием этого заболевания.
Во-первых, это ген белка-предшественника В-амилоида (АРР - Amyloid Precursor Protein), расположенный на 21 хромосоме и ответственный за 3 - 5% всех ранних (пресенильных) форм болезни Альцгеймера.
Во-вторых, это гены, расположенные на 14 и 1 хромосомах, обеспечивающие синтез мембранных белков - пресенелинов (PS1, пресенелин 1 и PS2, пресенелин 2). В нейронах головного мозга эти белки экспрессированы в эндоплазматическом ретикулуме тел нейронов и дендритов. При этом мутации гена PS1 обуславливают большинство (60 - 70%) всех ранних («семейных») форм болезни Альцгеймера, а мутации гена PS2, напротив, встречаются достаточно редко и достоверно известны только отдельные случаи заболевания в результате этих мутаций.
Разберем более подробно патогенез «мутационных» форм болезни Альцгеймера.
В начале следует разобраться в механизмах образования В-амилоида, так как со времени описания А.Альцгеймером заболевания, носящего его имя, и вплоть до относительно недавнего времени только образование В-амилоида, входящего в состав сенильных бляшек, считалось единственной причиной возникновения пресенильной деменции.
Следует указать, что продукция В-амилоида в незначительных количествах является результатом нормального процессинга белка, входящего в состав мембран нейронов и носящего уже известное нам название «белка предшественника В-амилоида» - АРР.
Образующийся В-амилоид - небольшой пептид, состоящий из 42 аминокислотных остатков, успешно метаболизируется и удаляется из головного мозга при посредстве глиальных клеток-чистильщиков - «скевенджеров» (схема 1). Полученные знания об особенностях метаболизма мембранного белка АРР позволили генетикам и патологам сделать несколько предположений о механизмах избыточного и вредоносного образования в головном мозгу В-амилоида.
Схема 1. Нормальный метаболизм Р-амилоида в нервной ткани
Во-первых, мутации гена АРР (в настоящее время известны 8 патогенных точечных мутаций этого гена) могут приводить к избыточному образованию В-амилоида, который не полностью выводится из организма и откладывается перицеллюлярно, образуя сенильные бляшки.
Во-вторых, процессинг АРР происходит под влиянием не нормальных, измененных ферментов-секретаз, за счет чего преимущественно образуются более длинные, склонные к фибриллярной агрегации формы В-амилоида
И, наконец, в-третьих, в накоплении В-амилоида «виновны» клетки-чистильщики, которые в силу каких либо причин не обладают достаточной активностью, или же их
количество просто недостаточно для успешной выполнении скевенджерной функции.
Исследования, проведенные относительно недавно, в 90-е годы прошлого столетия, показали, что из всех гипотез, объясняющих избыточное образование В-амилоида, основной является та, которая отдает приоритет патологическому процессингу АРР под влиянием мутантно измененных ферментов-секретаз.
Важным фактом для понимания механизмов патогенеза семейных форм болезни Альцгеймера явилось открытие ферментной активности уже упомянутых выше мембранных белков: пресенелина 1 и пресенелина 2. Иначе говоря, PSl и PS2 выполняют функции секретаз по отношению к мембранному белку АРР, осуществляя его процессинг до уровня В-амилоида. Секретазная активность пресенелинов по разному проявляется в отношении к различным сайтам белка АРР.
Это обстоятельство позволило определить и выделить несколько секретаз (альфа, бета и гамма секретазы), входящих в состав пресенелинов. Наиболее активными секретазами, ответственными за образование В-амилоида, оказались бета и гамма секретазы. Более того, именно бета и гамма секретазы обеспечивают образование удлиненных форм В-амилоида, обладающего повышенными агрегационными свойствами, что и приводит к возникновению перинейрональных сенильных бляшек. Установлено, что мутации в генах пресенелинов приводят к тому, что наиболее активным участком этого белка-фермента становится гамма-секретаза со всеми вытекающими отсюда патологическими для нейронов последствиями. Кроме того, было показано, что мутации генов пресенели- нов способны оказывать и ряд других, не связанных с ферментной активностью PS1 и PS2, патологических влияний на нейроны головного мозга, нарушая синаптогенез и, тем самым, разрушая межнейрональные контакты.Патогенная функция В-амилоида не ограничивается образованием сенильных бляшек. Установлено, что В-амилоид повышает активность фермента (киназы) гликоген- синтетазы-3 значительно усиливающей фосфорилирование тау-протеина, обеспечивающего стабильность микротрубочек нейронов. Гиперфосфорилированный тау-протеин теряет свои свойства, микротрубочки разрушаются и в нейронах на базе аномально сдвоенных филаментов измененного тау-протеина образуются нейрофибриллярные сплетения (нейрофибриллярные клубки) - схема 2.
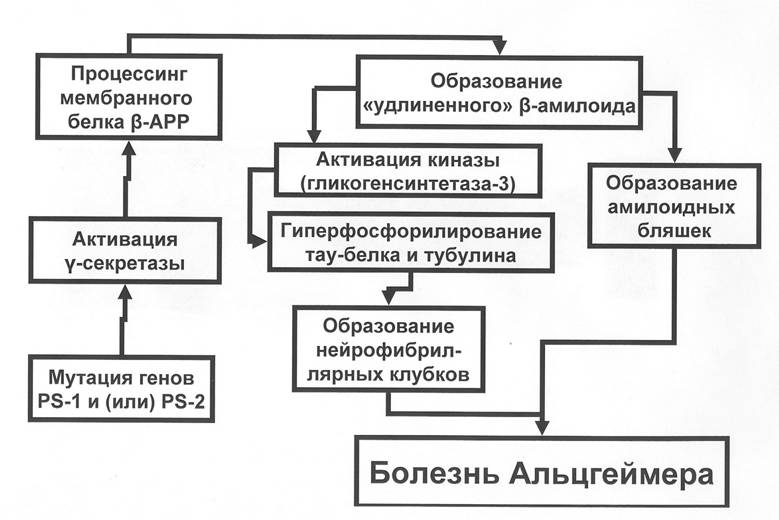
Схема 2. Механизмы формирования патологического Р-амилоида и нейрофибриллярных клубков
Таким образом, образование сенильных бляшек и нейрофибриллярных включений
- это не два раздельных патогенетических механизма, лежащих в основе возникновения и развития болезни Альцгеймера, но единый патологический процесс, запуск которого обеспечивается образованием аномального В-амилоида.
Обсуждая патогенетические механизмы болезни Альцгеймера, следует иметь в виду, что за исключением явных «семейных» форм этого заболевания, в остальных случаях специфический патологический процесс неизбежно, в силу возраста пациентов, развивается на фоне действия на головной мозг ряда неспецифических патологических факторов, например, таких как атеросклероз сосудов головного мозга, недостаточность антиоксидантных систем мозга, микровоспалительные очаги и т.п..
В приводимой здесь схеме (схема 3) сделана попытка объединить как генетически обусловленные патогенетические механизмы, лежащие в основе болезни Альцгеймера, так и факторы патогенеза этого заболевания, обусловленные возрастными изменениями соответствующих нервных структур.
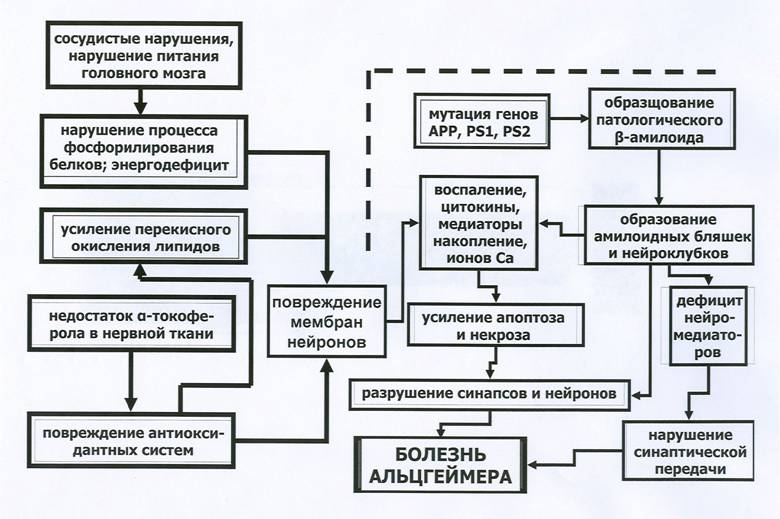
Схема 3. Основные патогенетические механизмы болезни Альцгеймера
Принципы патогенетической терапии болезни Альцгеймера
К сожалению, в настоящее время не разработано терапевтических методов и схем, позволяющих излечивать или, по крайней мере, остановить развитие болезни Альцгеймера. Применяемые немедикаментозные и медикаментозные методы лечения способны лишь замедлить течение этого тяжелого заболевания.
Все лекарственные средства, применяемые для облегчения состояния больных, находящихся на различных стадиях развития болезни Альцгеймера, можно разделить на несколько групп:
- препараты, замедляющие образование бета-амилоида (например, недавно введенный в практику препарат фенсерин, который помимо антиамилоидного действия обладает еще и свойством блокатора ацетилхолинэстеразы);
- препараты, предупреждающие дефицит нейромедиаторов в ЦНС: холиномиметические и адреномиметические препараты, ингибиторы ацетилхолинэстеразы. К этой же группе следует отнести и препараты ингибирующие обратный захват серотонина в соответствующих рецепторах;
- препараты, улучшающие психическое состояние больных: антидепрессанты, транквилизаторы;
- препараты, улучшающие кровоснабжение головного мозга (вазодилататоры), а также препараты, препятствующие развитию атеросклероза (например, статины);
- препараты, относящиеся к группе нейротрофиков и нейропртекторов (например, церебролизин, актовегии, различные антиоксиданты).
Средства немедикаментозной терапии болезни Альцгеймера должны быть направлены на улучшение психического и соматического состояния пациентов. Больных нельзя лишать общения с близкими им людьми, необходимо бороться с гиподинамией, продукты, входящие в состав диеты больных, должны быть легкоусвояемые и не содержать веществ, возбуждающих ЦНС.
II.
Еще по теме Болезнь Альцгеймера:
- Документа как объект криминалистического исследования, правила обращения с документами.
- План лекции
- Определение и краткая характеристика нейродегенеративных заболеваний
- Болезнь Альцгеймера
- Болезнь Паркинсона. Паркинсонизм
- Прионные нейродегенеративные заболевания
- Приложение
- Типы передачи наследственных болезней, характеристика, примеры.
- ТЕСТОВЫЕ ЗАДАНИЯ
- Роль усиления или ослабления апоптоза в развитии патологических процессов
- Апоптоз при нейродегенеративных заболеваниях
- § 2. Характеристика элементов, образующих уголовно-правовой ме- ханизм охраны прав и свобод пациента